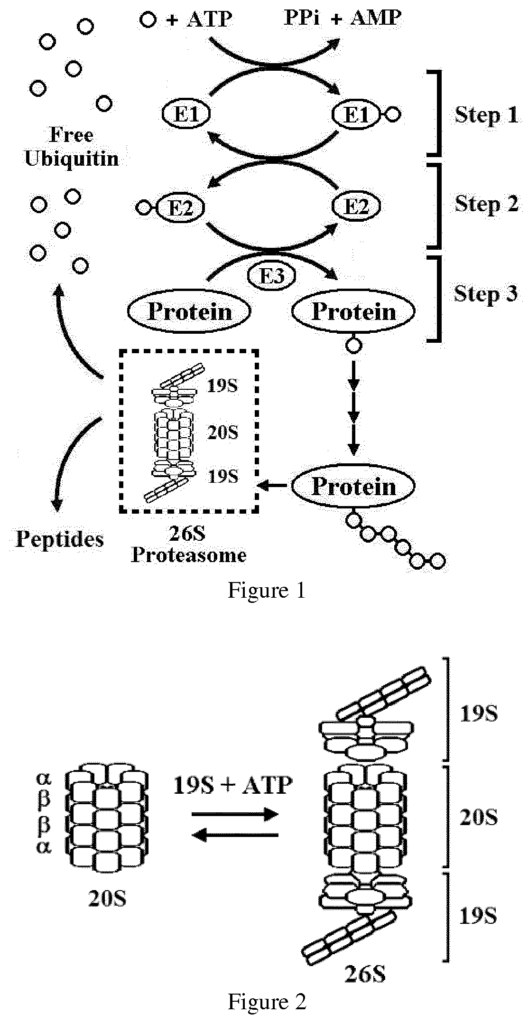
The present invention relates generally to proteasome inhibiting β-lactam compounds useful for the treatment of cancer and neurodegenerative disorders. The invention also provides pharmaceutical compositions and extended release formulations of said compounds, and medical uses of said compounds and/or pharmaceutical compositions to treat cancer and neurodegenerative disorders.
The invention claimed is:
1. A simonorealide compound of general formula (1a) or (1b):

wherein
X is H or —CO—O—R7;
Y is NR8, S(O)n, Se(O)n or Te(O)n;
R1 is selected from hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkyl, arylalkenyl, arylalkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, and substituted derivatives thereof;
R2 is —CH(OH)R9, —SO2—R, —SeO2—R or —TeO2—R with R is an alkyl, and substituted derivatives thereof;
R3 and R4 are independently selected from hydrogen, alkyl, halogen, haloalkyl, haloalkenyl, haloalkynyl, hydroxyalkyl, alkyl substituted by group selected from —O—SO2—R′, —O—SeO2—R′ or —O—TeO2—R′ with R′ is an alkyl, or an aryl, and substituted derivatives thereof;
R5 is selected from hydrogen, hydroxyl, sulfhydryl, alkyl, alkenyl, alkynyl, aryl, arylalkyl, arylalkenyl, arylalkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, heteroaryl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, cycloheteroalkyl, cycloheteroalkenyl, cycloheteroalkynyl, acyl, and substituted derivatives thereof;
R6 is selected from hydrogen, alkyl, halogen, haloalkyl, haloalkenyl, haloalkynyl, hydroxyalkyl, alkyl substituted by group selected from —O—SO2—R′, —O—SeO2—R′ or —O—TeO2—R′ with R′ is an alkyl, or an aryl, and substituted derivatives thereof;
R7 is —[C(R10)(R11)—O—CO]m—R12;
R8 is hydrogen, alkyl, and substituted derivatives thereof;
R9 is selected from hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkyl, arylalkenyl, arylalkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, heteroaryl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, cycloheteroalkyl, cycloheteroalkenyl, cycloheteroalkynyl, acyl, and substituted derivatives thereof;
R10 and R11 are independently selected from hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkyl, arylalkenyl, arylalkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, heteroaryl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, alkoxy, alkoxycarbonyl, carbamoyl, and substituted derivatives thereof;
R12 is selected from alkyl, alkenyl, alkynyl, aryl, arylalkyl, arylalkenyl, arylalkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, heteroaryl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, cycloheteroalkyl, cycloheteroalkenyl, cycloheteroalkynyl, acyl, and substituted derivatives thereof; or R12 is transport substrate of LAT1 selected from leucine, isoleucine, valine, phenylalanine, tyrosine, tryptophan, histidine, methionine, L-dopa, 3-O-methyldopa, α-methyl-phenylalanine, α-methyl-tyrosine, α-methyldopa, gabapentin, 3-iodo-α-methyl-L-tyrosine, 3-fluoro-α-methyl-L-tyrosine, 3-iodo-O-methyl-L-tyrosine, 3-iodo-O-methyl-α-methyl-L-tyrosine, 4-iodo-L-meta-tyrosine, 6-iodo-L-meta-tyrosine, O-(2-fluoroethyl)-L-tyrosine, 3-O-methyl-6-fluoro-L-dopa, 2-iodo-L-tyrosine, 2-fluoro-L-tyrosine, and substituted derivatives thereof; or R12 is a transport substrate of GLUT1, MCT1, CAT1, CNT2 or SVCT2; or R12 is a peptide substrate; or R12 is a peptide vector;
n is 0, 1 or 2;
m is 0 or 1;
with the proviso that when Y is NH then X is not H.
2. A compound according to claim 1, wherein
X is —CO—O—R12 or CO—O—C(R10)(R11)—O—CO—R12;
R10 and R11 are independently selected from hydrogen and alkyl; and
R12 is selected from alkyl, alkenyl, alkynyl, aryl, arylalkyl, arylalkenyl, arylalkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, heteroaryl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, cycloheteroalkyl, cycloheteroalkenyl, cycloheteroalkynyl, acyl, and substituted derivatives thereof, or R12 is transport substrate of LAT1 selected from leucine, isoleucine, valine, phenylalanine, tyrosine, tryptophan, histidine, methionine, L-dopa, 3-O-methyldopa, α-methyl-phenylalanine, α-methyl-tyrosine, α-methyldopa, gabapentin, 3-iodo-α-methyl-L-tyrosine, 3-fluoro-α-methyl-L-tyrosine, 3-iodo-O-methyl-L-tyrosine, 3-iodo-O-methyl-α-methyl-L-tyrosine, 4-iodo-L-meta-tyrosine, 6-iodo-L-meta-tyrosine, O-(2-fluoroethyl)-L-tyrosine, 3-O-methyl-6-fluoro-L-dopa, 2-iodo-L-tyrosine, 2-fluoro-L-tyrosine, and substituted derivatives thereof; or R12 is a transport substrate of GLUT1, MCT1, CAT1, CNT2 or SVCT2; or R12 is a peptide substrate; or R12 is a peptide vector.
3. A compound according to claim 1, wherein said compound corresponds to a general formula (2c), (2d), (3c), (3d), (4c), (4d), (5c) or (5d)

wherein R1, R2, R3, R4, R5, R6, R8, R9, R10, R11, and n are as defined in claim 1; and
R12 is selected from alkyl, alkenyl, alkynyl, aryl, arylalkyl, arylalkenyl, arylalkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, heteroaryl heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, cycloheteroalkyl, cycloheteroalkenyl, cycloheteroalkynyl, acyl, and substituted derivatives thereof.
4. A compound according to claim 1, wherein said compound corresponds to a general formula (2c), (2d), (3c), (3d), (4c), (4d), (5c) or (5d)

wherein R1, R2, R3, R4, R5, R6, R8, R9, R10, R11, and n are as defined herein; and
R12 is a transport substrate of LAT1 selected from leucine, isoleucine, valine, phenylalanine, tyrosine, tryptophan, histidine, methionine, L-dopa, 3-O-methyldopa, α-methyl-phenylalanine, α-methyl-tyrosine, α-methyldopa, gabapentin, 3-iodo-α-methyl-L-tyrosine, 3-fluoro-α-methyl-L-tyrosine, 3-iodo-O-methyl-L-tyrosine, 3-iodo-O-α-methyl-L-tyrosine, 4-iodo-L-meta-tyrosine, 6-iodo-L-meta-tyrosine, O-(2-fluoroethyl)-L-tyrosine, 3-O-methyl-6-fluoro-L-dopa, 2-iodo-L-tyrosine, 2-fluoro-L-tyrosine, and substituted derivatives thereof; or R12 is a transport substrate of GLUT1, MCT1, CAT1, CNT2 or SVCT2; or R12 is a peptide substrate; or R12 is a peptide vector.
5. A compound according to claim 1, wherein said compound corresponds to a general formula (2e), (2f), (3e), (3f), (4e), (4f), (5e) or (5f)

wherein R1, R2, R3, R4, R5, R6, R8, R9, and n are as defined in claim 1; and
R12 is a transport substrate of LAT1 selected from leucine, isoleucine, valine, phenylalanine, tyrosine, tryptophan, histidine, methionine, L-dopa, 3-O-methyldopa, α-methyl-phenylalanine, α-methyl-tyrosine, α-methyldopa, gabapentin, 3-iodo-α-methyl-L-tyrosine, 3-fluoro-α-methyl-L-tyrosine, 3-iodo-O-methyl-L-tyrosine, 3-iodo-O-methyl-α-methyl-L-tyrosine, 4-iodo-L-meta-tyrosine, 6-iodo-L-meta-tyrosine, O-(2-fluoroethyl)-L-tyrosine, 3-O-methyl-6-fluoro-L-dopa, 2-iodo-L-tyrosine, 2-fluoro-L-tyrosine, and substituted derivatives thereof; or R12 is a transport substrate of GLUT1, MCT1, CAT1, CNT2 or SVCT2; or R12 is a peptide substrate; or R12 is a peptide vector.
6. A compound according to claim 1 wherein R1 is H.
7. A compound according claim 1, wherein
R2 is —CH(OH)R9, —SO2—R, —SeO2—R or —TeO2—R;
R is an alkyl and substituted derivatives thereof;
R9 is selected from alkyl and cycloalkenyl.
8. A compound according to claim 1, wherein
R3 and R4 are independently selected from hydrogen, alkyl and haloalkyl;
R5 is selected from H or alkyl; and
R6 is selected from alkyl or haloalkyl.
9. A compound according to claim 1, wherein R8 is H.
10. A compound according to claim 1, wherein R12 is selected from alkyl, tyrosine, 3-iodo-α-methyl-L-tyrosine, 3-fluoro-α-methyl-L-tyrosine, 3-iodo-O-methyl-L-tyrosine, 3-iodo-O-methyl-α-methyl-L-tyrosine, 4-iodo-L-meta-tyrosine, 6-iodo-L-meta-tyrosine, O-(2-fluoroethyl)-L-tyrosine, 3-O-methyl-6-fluoro-L-dopa or 2-iodo-L-tyrosine, 2-fluoro-L-tyrosine.
11. A compound according to claim 1, wherein
R1 is H;
R3 and R4 are independently selected from hydrogen, alkyl and haloalkyl;
R5 is selected from hydrogen or alkyl;
R6 is selected from alkyl or haloalkyl;
R8 is hydrogen;
R9 is selected from alkyl and cycloalkenyl;
R10 and R11 are independently selected from hydrogen and alkyl.
12. A compound according to claim 11, wherein
R1 is H;
R3 is H;
R4 is methyl or 2-chloroethyl;
R5 is selected from hydrogen or methyl;
R6 is selected from methyl or 2-chloroethyl;
R8 is hydrogen;
R9 is selected from isopropyl and cyclohex-2-enyl;
R10 and R11 are independently selected from hydrogen and methyl.
13. A composition comprising a therapeutically effective amount of a compound as defined in claim 1, and a pharmaceutically acceptable vehicle, carrier or diluent.
14. A process of preparation of compounds of general formula (1b) of claim 1, wherein in general formula (1b) R2 is —SO2—R, —SeO2—R or —TeO2—R with R is an alkyl; R6 is chloroethyl, and X is H; comprising the following steps:
the formation of alkenyl azetidinone by a Staudinger reaction between acyl chloride and imine;
the iodine-promoted cyclization of to form iodopenem adduct;
the Stille cross-coupling between and tributyl(vinyl)tin to form compound;
the reverse Wacker oxidation of the carbon-carbon double bond of compound to form aldehyde;
the reduction of aldehyde to form alcohol;
optionally the oxidation of alcohol to form the corresponding sulfoxyde, sulfone, selenoxide, selenone, telluroxide or tellurone;
the desilylation to form compound;
the chlorination of compound to form chloride; and
the deprotection of the nitrogen of the β-lactam ring of compound to form proteasome inhibitor;

15. A process of preparation of compounds of general formula (1b) of claim 1, wherein in general formula (1b) R2 is —CH(OH)R9; R6 is chloroethyl; Y is S(O)n, Se(O)n or Te(O)n and n is 0, 1 or 2; and X is H; comprising the following steps:
the formation of alkenyl azetidinone by a Staudinger reaction between acyl chloride and imine;
the iodine-promoted cyclization of to form iodopenem adduct;
the ozonolysis of the carbon-carbon double bond of compound to form aldehyde;
the addition of R9—ZnCl on the aldehyde to form alcohol;
the Stille cross-coupling between and tributyl(vinyl)tin to form compound;
the reverse Wacker oxidation of the carbon-carbon double bond of compound to form aldehyde;
the reduction of aldehyde to form alcohol;
optionally the oxidation of alcohol to form the corresponding sulfoxide, sulfone, selenoxide, selenone, telluroxide or tellurone;
the desilylation to form compound;
the chlorination of compound to form chloride; and
the deprotection of the nitrogen of the β-lactam ring of compound to form proteasome inhibitor;

16. A process of preparation of compounds of general formula (1b) of claim 1, wherein in general formula (1b) R2 is —SO2—R, —SeO2—R or —TeO2—R with R is an alkyl; R6 is methyl; and X is H; comprising the following steps:
the formation of alkenyl azetidinone by a Staudinger reaction between acyl chloride and imine;
the iodine-promoted cyclization of to form iodopenem adduct;
the cross-coupling between and methylmagnesium bromide to form compound;
optionally the oxidation of alcohol to form the corresponding sulfoxyde, sulfone, selenoxide, selenone, telluroxide or tellurone;
the desilylation to form compound; and
the deprotection of the nitrogen of the β-lactam ring of compound to form proteasome inhibitor;

17. A process of preparation of compounds of general formula (1b) of claim 1, wherein in general formula (1b) R2 is —CH(OH)R9; R6 is methyl; Y is S(O)n, Se(O)n or Te(O)n and n is 0, 1 or 2; and X is H; comprising the following steps:
the formation of alkenyl azetidinone by a Staudinger reaction between acyl chloride and imine;
the iodine-promoted cyclization of to form iodopenem adduct;
the ozonolysis of the carbon-carbon double bond of compound to form aldehyde;
the addition of R9—ZnCl on the aldehyde to form alcohol;
the cross-coupling between and methylmagnesium bromide to form compound;
optionally the oxidation of alcohol to form the corresponding sulfoxide, sulfone, selenoxide, selenone, telluroxide or tellurone;
the desilylation to form compound; and
the deprotection of the nitrogen of the β-lactam ring of compound to form proteasome inhibitor;

18. The method of claim 15, wherein n is 0.
19. The method of claim 17, wherein n is 0.